大体系GVB初始轨道的自动构造
正确描述分子从平衡位置到解离极限的势能面必须同时处理好静态相关和动态相关。在静态相关方面,参考态波函数的选取至关重要。传统的Hartree-Fock(HF)波函数对主簇元素化合物在平衡位置是一个较好的参考态,但对远离平衡的结构,双、多自由基以及过渡金属化合物则不尽人意。这些体系具有典型的多参考特征,需要用多组态/多参考波函数来描述。常用的完全活性空间自洽场方法(complete active space self-consistent-field, CASSCF)计算量随活性空间呈指数增长,只适用于小分子体系。另一类多参考波函数是广义价键(perfect-pairing generalized valence bond theory, GVB) 波函数。这种波函数被定义为一系列对函数(germinal)的张量积,每个geminal由一个成键轨道和对应的反键轨道构成;相比于HF, 它包含了部分的静态相关,可处理CASSCF难于处理的大分子体系,因此是大体系电子相关计算合适的多参考态波函数。然而,一个典型的GVB计算要求手动地选出活性轨道,并将其一一配对,对一个较大的、复杂的体系而言,这几乎是一个不可能完成的任务。 近日,我们课题组与美国Arkansas大学Peter Pulay教授合作提出了一种自动构造GVB初始轨道的有效算法,可以实现大体系GVB的常规计算。该算法根据一定策略(依赖于体系RHF的稳定性)选出活性轨道,局域化后利用Kuhn-Munkres配对算法完成配对。基于此算法,一般分子的GVB计算能够像HF计算一样简单易行。
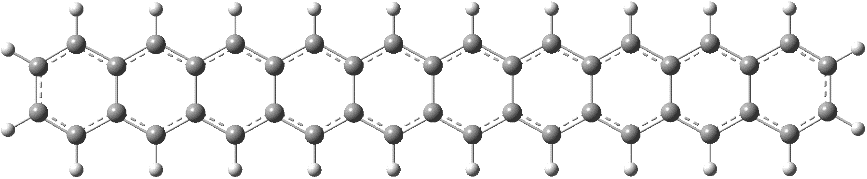
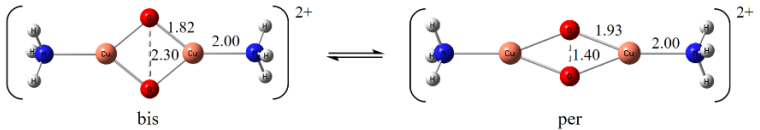
该工作中,我们应用此算法测试了一些实际体系(如图1所示),比如并苯(polyacene)体系和双核Cu化合物。对并苯体系,我们计算了垂直激发和绝热激发能,结果与实验值接近。其中涉及的最大并苯体系活性轨道多达96对,这是第一次报道活性空间超过50对的大体系GVB计算。对双核Cu化合物,在大活性空间的GVB计算完成后,我们可自动定义最小活性空间,并进行CASSCF-NEVPT2计算,对两个结构的稳定性的预测结果与其它大活性空间的多参考方法相似。基于GVB轨道自动定义最小活性空间,更加有效地处理大活性空间,可使多参考电子相关计算易于进行,这是此工作第二大亮点。
图1 文中研究的体系的体系
|