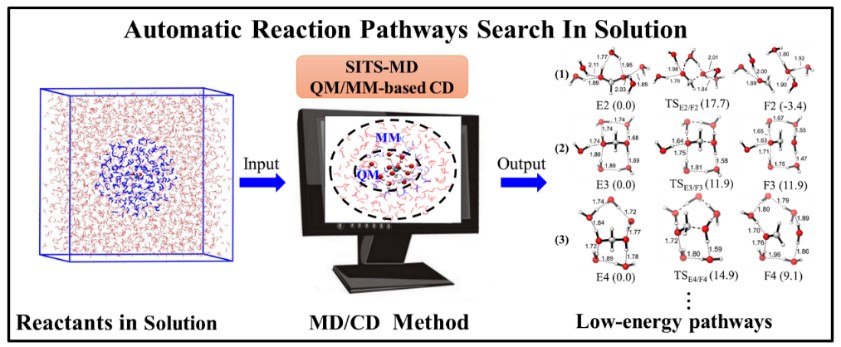
MD/CD方法:溶液中化学反应路径的自动搜索用
理论模拟在反应机理研究领域中发挥着非常重要的作用。通过计算模拟,人们可以解释反应机理,筛选理想的催化剂,以及预测和设计新反应等等。对于复杂的反应体系,尤其对于立体选择性高,分子柔性程度大的体系,通过人工方法搜索反应路径变得非常困难。 因此,发展自动化反应路径搜索方法是反应机理研究领域的热点课题。
2017年,本课题组发展了一种高效低成本的反应路径自动搜索方法:分子动力学模拟和反应坐标牵引相结合的方法(MD/CD)(J. Phys. Chem. A 2017, 121, 1351−1361)。在该方法中,采用基于分子力学(MM)或半经验量子力学方法(如PM6)的分子动力学(MD)模拟搜索极小值点的构象空间,改进的反应坐标牵引(CD)方法搜索可能的化学键形成或断裂的结构异构体以及对应的过渡态。
近日,我们通过对MD/CD方法的改进,将其推广到溶液中反应路径的自动搜索。在此工作中,为了更好的考虑溶剂对化学反应的影响,我们采用了选择性温度积分的增强抽样(SITS)MD模拟搜索反应物(或中间体,产物)在真实溶剂中的构象空间。此外,在CD计算中,我们采用显示溶剂模型(考虑>100 个溶剂分子)并结合QM/MM方法实现反应路径自动搜索。其中,反应物和 5~10个溶剂分子在QM区域,其余的溶剂分子在MM区域。
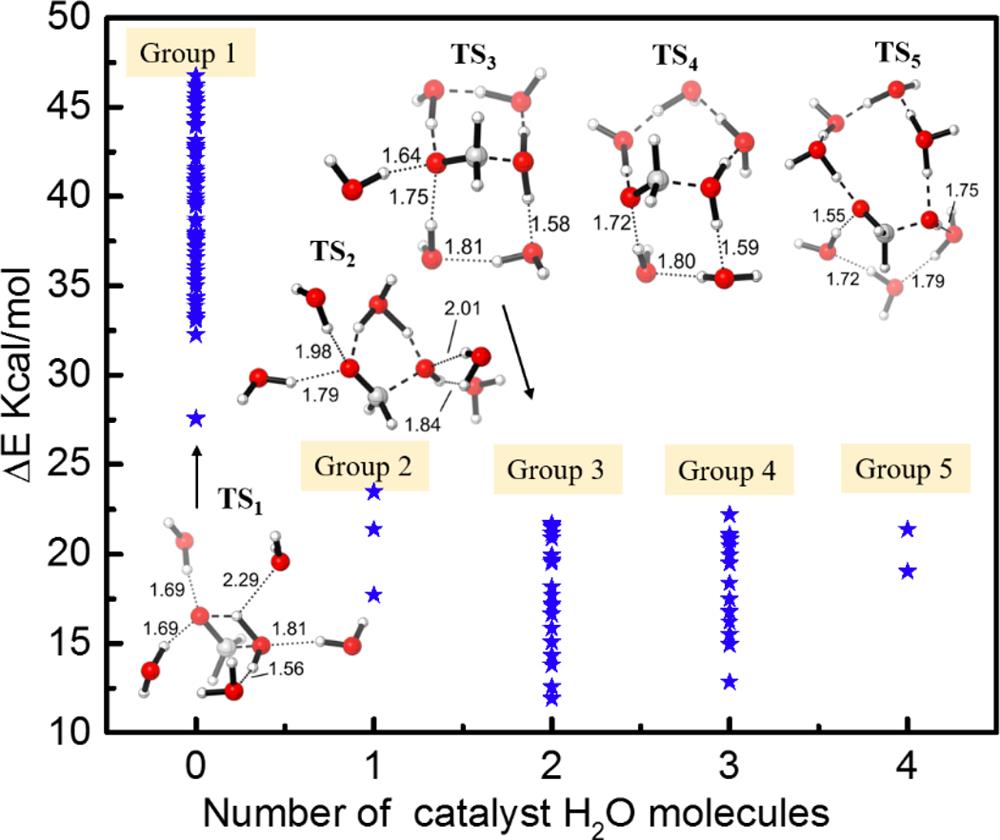
此工作中,该方法被应用于研究溶液中的化学反应:氧化胺在水和二甲亚砜(DMSO)溶液中的Cope消除反应;甲二醇在水溶液中的脱水反应。该方法可以自动搜索到一系列的低能反应路径。对于氧化胺的Cope消除反应,MD/CD结果可以定性地解释实验现象:氧化胺在 DMSO 溶液中反应更有利。 对于甲二醇的脱水反应,MD/CD 搜到 5 种不同的反应模式。基于其中的三种低能反应模式(1~3 个水分子直接参与催化),我们通过进一步的平均力势(PMF)计算确定了2 个水分子催化的水解路径最有利。MD/CD有望成为一种高效、计算经济的用于自动搜索溶液中化学反应路径的理论工具。
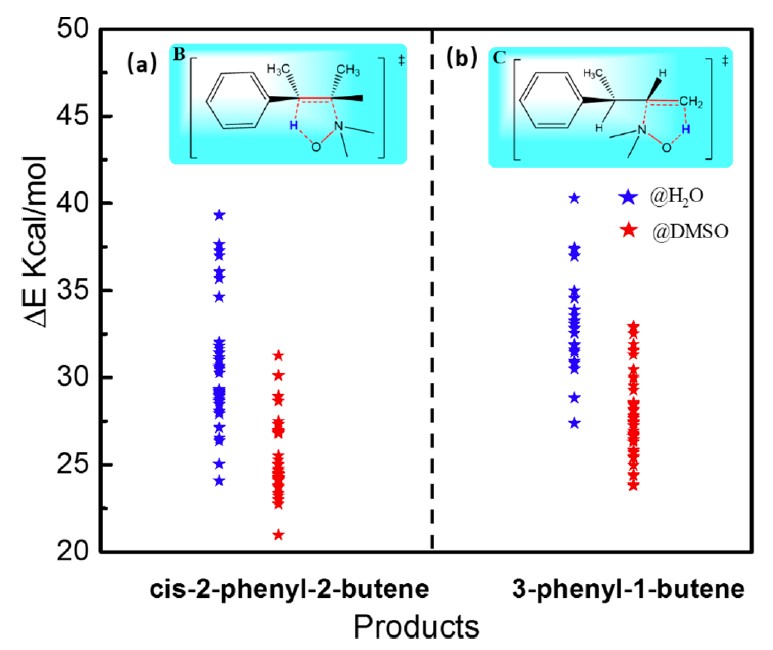 
氧化胺的Cope消除反应
甲二醇的脱水反应
|