普适的基于能量的分块方法计算大尺寸金属配位超分子的结构和性质
传统量子化学方法的计算量随着分子体系变大而迅速增大。分块方法是一种线性标度方法,能对较大的分子体系进行量子化学计算。分块方法的基本原理是将所要计算的目标体系分解为一系列较小的子体系,然后对这些子体系进行量子化学计算,最后将子体系的能量或性质组装来获得目标体系的能量或性质。自2005年至今,国际上发展了多种不同的分块方法,例如本课题组发展的普适的基于能量的分块方法(the generalized energy-based fragmentation, GEBF)和明尼苏达大学Truhlar教授课题组发展的多体展开方法(Electrostatically embedded many-body, EE-MB)等。目前分块方法广泛地应用于分子簇和共价键体系,然而利用分块方法处理含多个金属的配位超分子体系尚未见报道。
近期,本课题组把GEBF方法推广到计算大尺寸金属配位超分子的结构和性质。我们发现GEBF方法在计算配位化合物时必须采用特定的分块策略,即保留配位键,通过切断配体上的共价单键将大体系分为很多块,另外,子体系的基态自旋应通过各自的量子化学计算来确定。为了验证这一策略的可靠性,对一个中等尺寸配位超分子,发现GEBF方法得到的基态能量、结构、核磁(NMR)和红外光谱均和传统量子化学计算得到的相应结果很接近,证明按照上述分块策略GEBF方法能够准确描述配位超分子。
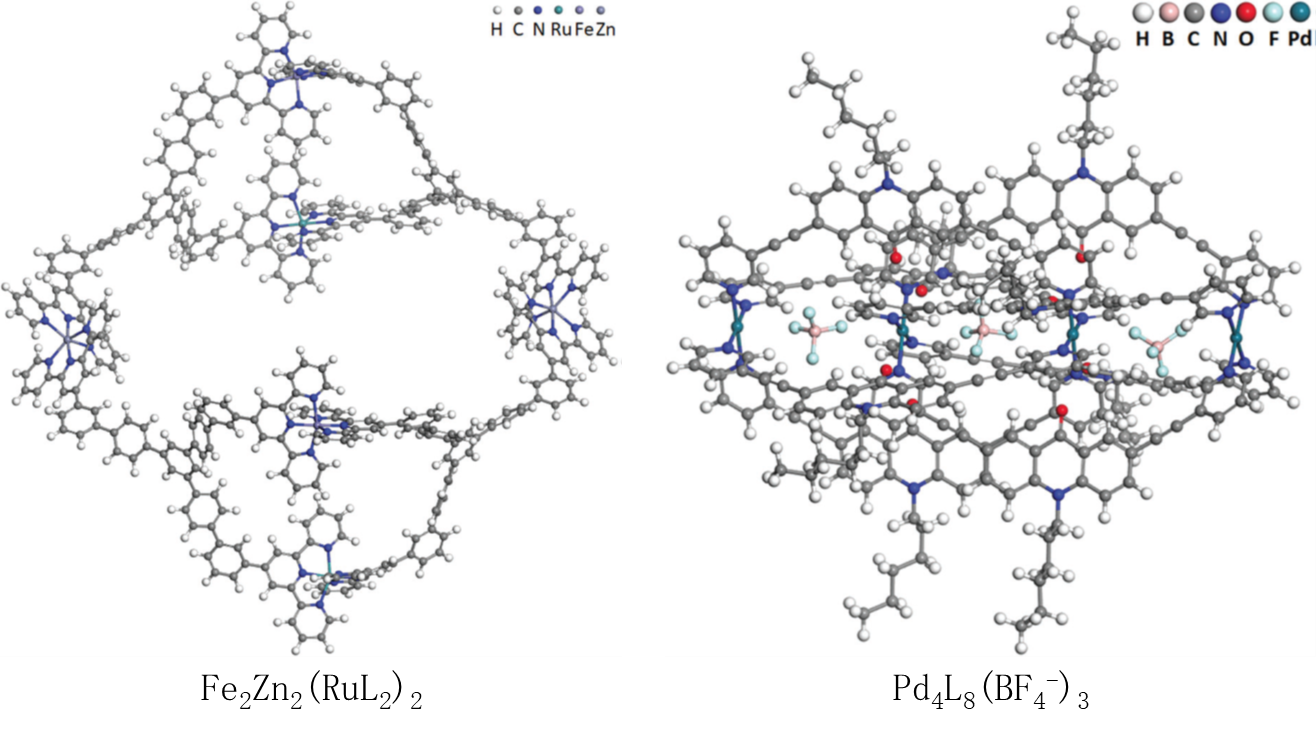
进一步地,将此策略应用于两个大尺寸金属配位超分子(见上图)。对含有618个原子的Fe2Zn2(RuL2)2超分子(如图)优化结构后计算1H核磁谱(共17140个基函数)。对主要的特征峰,GEBF计算得到的1H核磁谱和实验谱吻合,证明了GEBF计算可以揭示金属配位超分子的主要结构特征。对含有531个原子的Pd4L8(BF4-)3超分子主客体复合物,优化结构后计算红外光谱。GEBF计算得到红外光谱和实验谱吻合,特征峰频率差别小于30 cm-1。另外GEBF计算能帮助指认实验谱中对应的振动模式。
该工作进一步推广了GEBF方法的应用范围,为GEBF方法广泛地应用于金属配位分子领域奠定了基础。
|